2.2. 16s核糖体序列拼接实验¶
2.2.1. 1. 拼接思路¶
graph TD
A[swalign]-->B(匹配结果)
B-->C{寻找最长的相同片段}
C-->D[计算正反两条链在相同片段的各自部分]
D-->E[完成拼接]
2.2.2. 2.核心算法¶
算法的核心是利用swalign寻找到最长的相同片段。为此,我们开发的了一个函数Same_length
function [Res] = Same_length(seq)
% 本函数用于计算长度最长的重复序列,seq为Alignment的第二行,即由'|:-’组成的序列
% seq为序列,Res为包含最长连续片段的起点和终点组成的向量
% Copywrite by MTC, 2020.8.4, By Xia Menglei, Version 1.0
Res=(seq~='|');
po=find(Res==1);
po1=diff(po);
po2=find(po1==max(po1));
Res=[po(po2)+1,po(po2+1)-1];
end
Res返回的是共同序列在正链上的起始位点和终止位点。正义链是自己定的,一般公司会给出两条链,如果定义a为正,则b为负,反之亦然。只要保证全程都以该链为正即可。
2.2.3. 3.具体实例¶
以我们课题组之前的一次实验为案例进行说明
2.2.3.1. 3.1 数据读取¶
zheng='GCCATGCGGCGTGCTTACCATGCAGTCGAGCGGGGAACTTCGGTTCCCAGCGGCGGACGGGTGAGTAACACGTGGGTAACCTACCTCATAGTGGGGAATAGCCTTTCGAAAGGAAGATTAATACCGCATAATACTCGAGAATCGCATGATTCTTGAGCCAAAGGATTTATTCGCTATGAGATGGACCCGCGGCGCATTAGCTTGTTGGTGAGGTAACGGCTCACCAAGGCTTCGATGCGTAGCCGACCTGAGAGGGTGATCGGCCACATTGGAACTGAGACACGGTCCAGACTCCTACGGGAGGCAGCAGTGGGGAATATTGCACAATGGGGGAAACCCTGATGCAGCAACGCCGCGTGAGTGATGAAGGTCTTCGGATCGTAAAACTCTGTCTTATGGGACGATAATGACGGTACCATAGGAGGAAGCCACGGCTAACTACGTGCCAGCAGCCGCGGTAATACGTAGGTGGCAAGCGTTGTCCGGATTTACTGGGCGTAAAGGATGTGTAGGCGGATATTTAAGTGAGATGTGAAATCCCCGGGCTTAACTTGGGGGCTGCATTTCAAACTGGATGTCTGGAGTGCAGGAGAGGAAGGCAGAATTCCTAGTGTAGCGGTGAAATGCGTAGAGATTAGGAAGAATACCAGTGGCGAAGGCGGCCTTCTGGACTGTAACTGACGCTGAGGCATGAAAGCGTGGGGAGCAAACAGGATTAGATACCCTGGTAGTCCACGCCGTAAACGATGAATACTAGGTGTAGGAGGTATCGACTCCTTCTGTGCCGCAGTTAACACAATAAGTATTCCGCCTGGGAAGTACGGTCGCAAGATTAAAACTCAAAGGAATTGACGGGGACCCGCACAAGCAGCGGAGCATGTGGTTTAATTCGAAGCAACGCGAAGAACCTTACCTAGACTTGACATCTCCTGAATTAGTCCGTAATGGATGAAGTCCCTTTCGGGACAGGATGACAGGTGGTGCATGGTTGTCGTCAGCTCGTGTCGTGAGATGTTGGGTTAAGTCCCGCACGAGCGCACCCTATCATAGTGCTAACATTAGTGAGCACTCTAGTGAGACTGCCGGTACGGGAGAAGGTGGGATGACGTCCAATTCATTCATTGCCCCTTATGTTCTAG';
fan='GCCCCCTCTTCGACCGCTGGTTCCAAAAGGTTACCTCACGGGCTTCGGGTGTTGCCGACTCTCATGGTGTGACGGGCGGTGTGTACAAGACCCGGGAACGTATTCACCGCGACATTCTGATTCGCGATTACTAGCAACTCCGGCTTCATGTAGGCGAGTTTCAGCCTACAATCCGAACTGGGATGGGGTTTTGAGTTTTGCTCCACCTTGCGGTATTGCATCTTTTTGTCCCCACCATTGTAGCACGTGTGTAGCCCTAGACATAAGGGGCATGATGATTTGACGTCATCCCCACCTTCCTCCCGGTTAACCCGGGCAGTCTCACTAGAGTGCTCAACTAAATGTTAGCAACTAATGATAAGGGTTGCGCTCGTTGCGGGACTTAACCCAACATCTCACGACACGAGCTGACGACAACCATGCACCACCTGTCATCCTGTCCCCGAAGGGACTTCATCCATTACGGACTAATTCAGGAGATGTCAAGTCTAGGTAAGGTTCTTCGCGTTGCTTCGAATTAAACCACATGCTCCGCTGCTTGTGCGGGTCCCCGTCAATTCCTTTGAGTTTTAATCTTGCGACCGTACTTCCCAGGCGGAATACTTATTGTGTTAACTGCGGCACAGAAGGAGTCGATACCTCCTACACCTAGTATTCATCGTTTACGGCGTGGACTACCAGGGTATCTAATCCTGTTTGCTCCCCACGCTTTCATGCCTCAGCGTCAGTTACAGTCCAGAAGGCCGCCTTCGCCACTGGTATTCTTCCTAATCTCTACGCATTTCACCGCTACACTAGGAATTCTGCCTTCCTCTCCTGCACTCCAGACATCCAGTTTGAAATGCAGCCCCCAAGTTAAGCCCGGGGATTTCACATCTCACTTAAATATCCGCCTACACATCCCTTTACGCCCAGTAAATCCGGACACGCTTGCCACCTACGTATTACCGCGGCTGCTGGCACGTAGTTAGCGTGCTTCCTCCTATGGTACCGTCATTATCGTCCATAAGACAGAGTTTACGATCCGAAGACCTTCATCACTCACGCGCGTGCTGCATCAGGCTTTCCCCAATGGTGCATATTCCCATGCTGCCTCCCGTAGGATCTGGAACGGGTCTCAGTTTCCAATGGGGGCGAATACCCCTCTCTCAGGGTCGGGCTTA';
2.2.3.2. 3.2 进行swalign,确定最长的相同部分¶
fan1=seqrcomplement(fan);
[Score, Alignment] = swalign(zheng, fan1);
t=Alignment(2,:);
Res = Same_length(t);
Alignment1=Alignment(1,:);
same_seq=Alignment1(Res(1):Res(2));
需要说明的是: - Same_length为自己开发的函数,该函数可以判断出Alignment中最长的相同序列,并返回该序列在zheng序列的起始和终止位置(Res); - same_seq为最长的相同序列(以zheng为模板)
2.2.3.3. 3.3 进行序列拼接¶
po1=strfind(zheng,same_seq);
po2=strfind(fan,seqrcomplement(same_seq));
seq_total=[zheng(1:po1-1),same_seq,seqrcomplement(fan(1:po2-1))];
seq_total即为拼接的结果
2.2.4. 4. 进行结果验证¶
2.2.4.1. 4.1 直接与公司结果作比较¶
a='CTGGTTCCAAAGGTTACCTCACGGGCTTCGGGTGTTGCCGACTCTCATGGTGTGACGGGCGGTGTGTACAAGACCCGGGAACGTATTCACCGCGACATTCTGATTCGCGATTACTAGCAACTCCGGCTTCATGTAGGCGAGTTTCAGCCTACAATCCGAACTGGGATGGGGTTTTGAGTTTTGCTCCACCTTGCGGTATTGCATCTTTTTGTCCCCACCATTGTAGCACGTGTGTAGCCCTAGACATAAGGGGCATGATGATTTGACGTCATCCCCACCTTCCTCCCGGTTAACCCGGGCAGTCTCACTAGAGTGCTCAACTAAATGTTAGCAACTAATGATAAGGGTTGCGCTCGTTGCGGGACTTAACCCAACATCTCACGACACGAGCTGACGACAACCATGCACCACCTGTCATCCTGTCCCCGAAGGGACTTCATCCATTACGGACTAATTCAGGAGATGTCAAGTCTAGGTAAGGTTCTTCGCGTTGCTTCGAATTAAACCACATGCTCCGCTGCTTGTGCGGGTCCCCGTCAATTCCTTTGAGTTTTAATCTTGCGACCGTACTTCCCAGGCGGAATACTTATTGTGTTAACTGCGGCACAGAAGGAGTCGATACCTCCTACACCTAGTATTCATCGTTTACGGCGTGGACTACCAGGGTATCTAATCCTGTTTGCTCCCCACGCTTTCATGCCTCAGCGTCAGTTACAGTCCAGAAGGCCGCCTTCGCCACTGGTATTCTTCCTAATCTCTACGCATTTCACCGCTACACTAGGAATTCTGCCTTCCTCTCCTGCACTCCAGACATCCAGTTTGAAATGCAGCCCCCAAGTTAAGCCCGGGGATTTCACATCTCACTTAAATATCCGCCTACACATCCTTTACGCCCAGTAAATCCGGACAACGCTTGCCACCTACGTATTACCGCGGCTGCTGGCACGTAGTTAGCCGTGGCTTCCTCCTATGGTACCGTCATTATCGTCCCATAAGACAGAGTTTTACGATCCGAAGACCTTCATCACTCACGCGGCGTTGCTGCATCAGGGTTTCCCCCATTGTGCAATATTCCCCACTGCTGCCTCCCGTAGGAGTCTGGACCGTGTCTCAGTTCCAATGTGGCCGATCACCCTCTCAGGTCGGCTACGCATCGAAGCCTTGGTGAGCCGTTACCTCACCAACAAGCTAATGCGCCGCGGGTCCATCTCATAGCGAATAAATCCTTTGGCTCAAGAATCATGCGATTCTCGAGTATTATGCGGTATTAATCTTCCTTTCGAAAGGCTATTCCCCACTATGAGGTAGGTTACCCACGTGTTACTCACCCGTCCGCCGCTGGGAACCGAAGTTCCCCGCTCGACTGC';
%a为公司给出的拼接结果
[~,Alignment]=nwalign(seq_total,seqrcomplement(a));
seqalignviewer(Alignment);
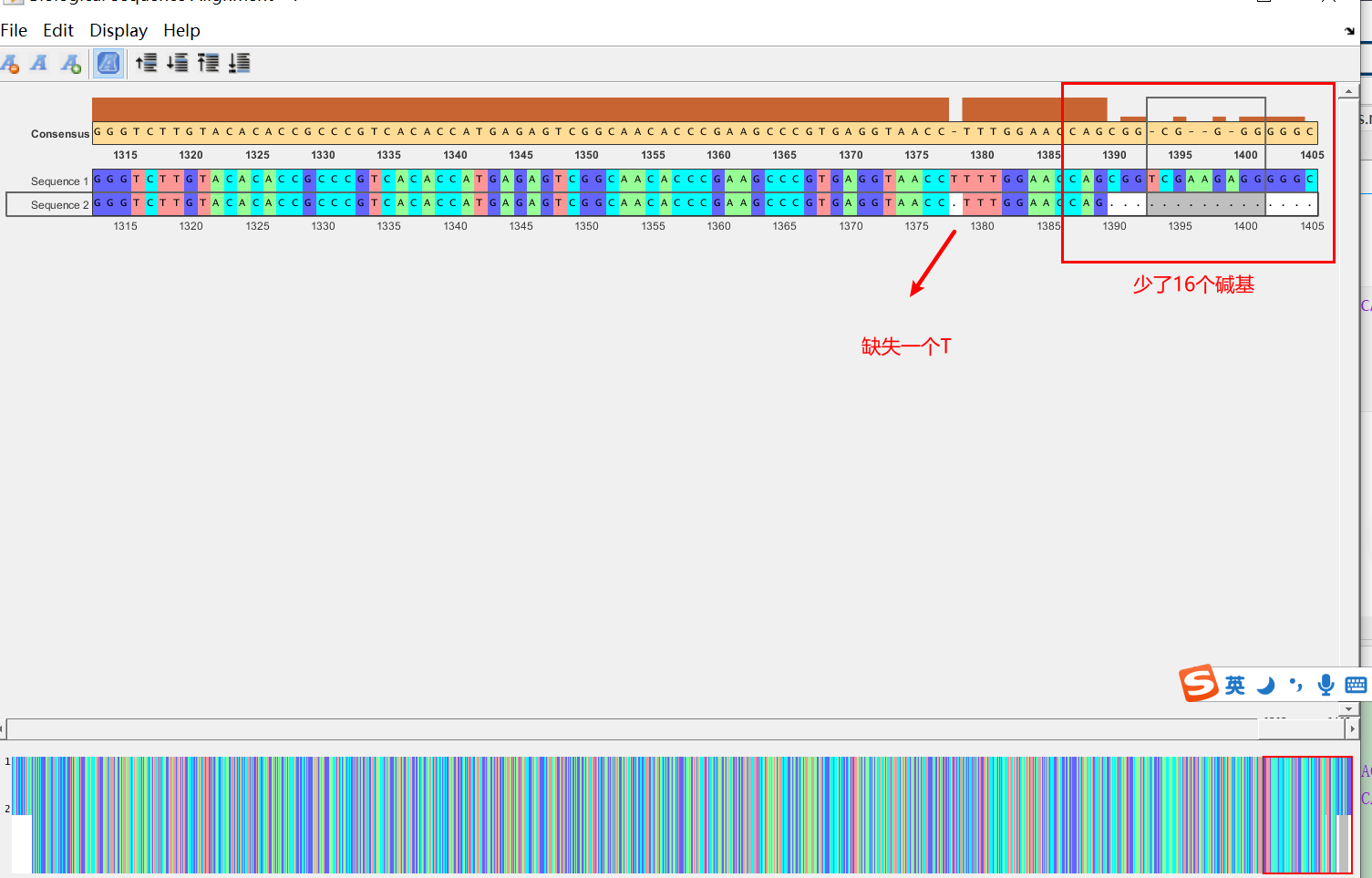
- a为公司拼接的结果,seq_total为使用我们的算法计算出的结果
- 可以看出,公司给出的拼接结果少了一个T,并且两端均少了很多碱基(如右边减少了16个碱基)
2.2.4.2. 4.2 利用ATCC 824序列进行比对¶
fasta_download('NR_074511.2');
seq=fastaread(['NR_074511.2','.fasta']).Sequence
[Score1,Alignment1]=nwalign(seq_total,seq);
seqalignviewer(Alignment1)
[Score2,Alignment2]=nwalign(seqrcomplement(a),seq);
seqalignviewer(Alignment2)
说明:
- fast_download为我们开发的序列文件自动下载程序,见:https://mtc.readthedocs.io/en/latest/Gene/Primer_blast.html#efetchncbi
- 通过对比可以返现,我们的拼接结果优于公司结果,但是该序列仍旧与MTCC 824的16s序列有差异;
- 公司对上述差异给出的解释是:16s测序时,当开始的前30个序列检测是不准确的,因此在分析数据是,会直接将两条链的前30左右碱基去掉
用户留言