2.3. Dna Features Viewer¶
2.3.1. 方法说明¶
pip install dna_features_viewer
pip install bokeh pandas
pip install bcbio-gff
2.3.2. 用法说明书¶
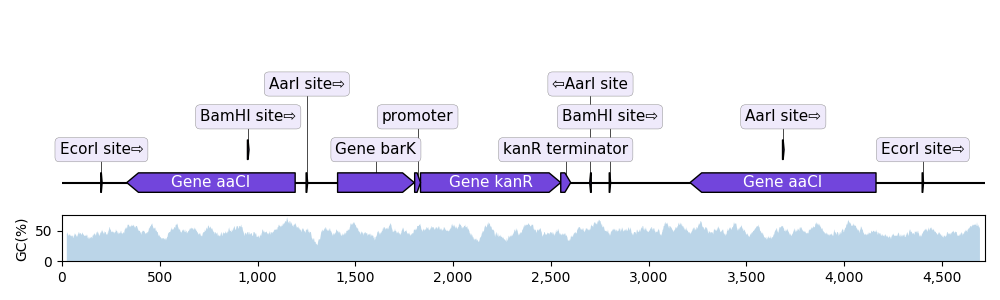
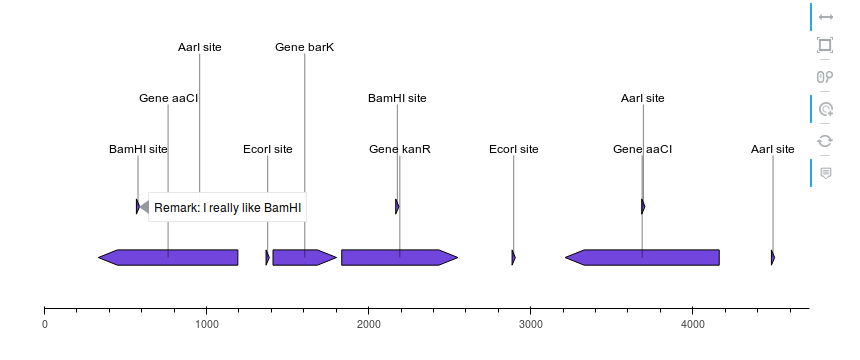
2.3.2.1. with_gc_plot¶
"""In this example we plot a record's annotations on top of the curve of the
local GC content in the record's sequence.
"""
import matplotlib.pyplot as plt
from dna_features_viewer import BiopythonTranslator
from Bio import SeqIO
import numpy as np
def plot_local_gc_content(record, window_size, ax):
"""Plot windowed GC content on a designated Matplotlib ax."""
def gc_content(s):
return 100.0 * len([c for c in s if c in "GC"]) / len(s)
yy = [
gc_content(record.seq[i : i + window_size])
for i in range(len(record.seq) - window_size)
]
xx = np.arange(len(record.seq) - window_size) + 25
ax.fill_between(xx, yy, alpha=0.3)
ax.set_ylim(bottom=0)
ax.set_ylabel("GC(%)")
record = SeqIO.read("example_sequence.gb", "genbank")
translator = BiopythonTranslator()
graphic_record = translator.translate_record(record)
fig, (ax1, ax2) = plt.subplots(
2, 1, figsize=(10, 3), sharex=True, gridspec_kw={"height_ratios": [4, 1]}
)
graphic_record.plot(ax=ax1, with_ruler=False, strand_in_label_threshold=4)
plot_local_gc_content(record, window_size=50, ax=ax2)
fig.tight_layout() # Resize the figure to the right height
fig.savefig("with_gc_plot.png")
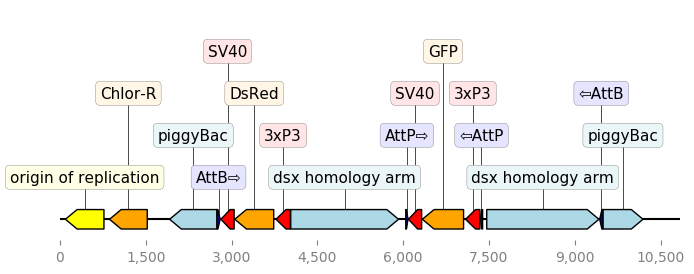
2.3.2.2. translator_with_cutom_colors¶
"""An example with GIF generation at the end. How cool is that!
This example requires the Moviepy library installed (pip install moviepy).
"""
from Bio import Entrez, SeqIO
from dna_features_viewer import BiopythonTranslator
# DOWNLOAD THE PLASMID's RECORD FROM NCBI
Entrez.email = "zulko@egf.org"
handle = Entrez.efetch(
db="nucleotide", id=1473096477, rettype="gb", retmode="text"
)
record = SeqIO.read(handle, "genbank")
# CREATE THE GRAPHIC RECORD WITH DNA_FEATURES_VIEWER
color_map = {
"rep_origin": "yellow",
"CDS": "orange",
"regulatory": "red",
"misc_recomb": "darkblue",
"misc_feature": "lightblue",
}
translator = BiopythonTranslator(
features_filters=(lambda f: f.type not in ["gene", "source"],),
features_properties=lambda f: {"color": color_map.get(f.type, "white")},
)
translator.max_line_length = 15
graphic_record = translator.translate_record(record)
ax, _ = graphic_record.plot(figure_width=8, strand_in_label_threshold=7)
ax.figure.savefig("translator_with_custom_colors.png", bbox_inches="tight")
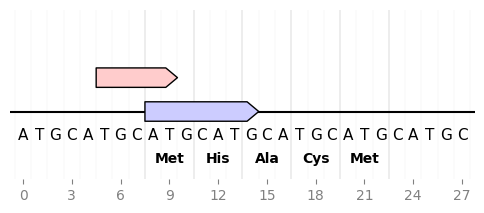
2.3.2.3. sequence_and_translation¶
from dna_features_viewer import GraphicFeature, GraphicRecord
record = GraphicRecord(sequence="ATGCATGCATGCATGCATGCATGCATGC", features=[
GraphicFeature(start=5, end=10, strand=+1, color='#ffcccc'),
GraphicFeature(start=8, end=15, strand=+1, color='#ccccff')
])
ax, _ = record.plot(figure_width=6)
record.plot_sequence(ax, guides_intensity=0.2)
fontdict = {'weight': 'bold'}
record.plot_translation(ax, (8, 23), fontdict=fontdict, guides_intensity=0.8)
ax.figure.savefig('sequence_and_translation.png', bbox_inches='tight')
2.3.2.4. plot_with_bokeh¶
"""Simple example with Bokeh output. Requires the Bokeh library installed.
"""
from dna_features_viewer import BiopythonTranslator
from bokeh.resources import CDN
from bokeh.embed import file_html
record = BiopythonTranslator().translate_record(record="example_sequence.gb")
plot = record.plot_with_bokeh(figure_width=8)
with open("plot_with_bokeh.html", "w+") as f:
f.write(file_html(plot, CDN, "Example Sequence"))
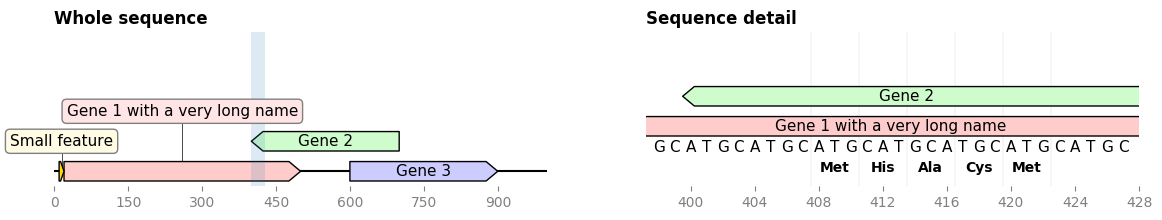
2.3.2.5. overview_and_detail¶
"""Generate a 2-plot figure with full sequence on the left,
detail of a sub-segment on the right."""
from dna_features_viewer import GraphicFeature, GraphicRecord
import matplotlib.pyplot as plt
record = GraphicRecord(sequence=250 * "ATGC", features=[
GraphicFeature(start=10, end=20, strand=+1, color="#ffd700",
label="Small feature"),
GraphicFeature(start=20, end=500, strand=+1, color="#ffcccc",
label="Gene 1 with a very long name"),
GraphicFeature(start=400, end=700, strand=-1, color="#cffccc",
label="Gene 2"),
GraphicFeature(start=600, end=900, strand=+1, color="#ccccff",
label="Gene 3")
])
zoom_start, zoom_end = 398, 428 # coordinates of the "detail"
cropped_record = record.crop((zoom_start, zoom_end))
fig, (ax1, ax2) = plt.subplots(1, 2, figsize=(14, 2))
# PLOT THE WHOLE SEQUENCE
ax1.set_title("Whole sequence", loc='left', weight='bold')
record.plot(ax=ax1)
ax1.fill_between((zoom_start, zoom_end), +1000, -1000, alpha=0.15)
# PLOT THE SEQUENCE DETAILS
cropped_record.plot_translation(ax=ax2, location=(408, 423),
fontdict={'weight': 'bold'})
cropped_record.plot(ax=ax2, plot_sequence=True)
ax2.set_title("Sequence detail", loc='left', weight='bold')
fig.savefig('overview_and_detail.png', bbox_inches='tight')
2.3.2.6. multipage_plot¶
"""In this example we plot a record fragment with sequence over multiple lines.
"""
from dna_features_viewer import BiopythonTranslator
class CustomTranslator(BiopythonTranslator):
def compute_feature_color(self, feature):
return {
"restriction_site": "yellow",
"CDS": "orange",
"promoter": "darkblue",
"terminator": "lightblue",
}[feature.type]
translator = CustomTranslator()
graphic_record = translator.translate_record("example_sequence.gb")
subrecord = graphic_record.crop((1800, 2750))
subrecord.plot_on_multiple_pages(
"multipage_plot.pdf",
nucl_per_line=70,
lines_per_page=7,
plot_sequence=True,
)


2.3.2.7. multiline_plot¶
"""In this example we plot a record fragment with sequence over multiple lines.
"""
from dna_features_viewer import BiopythonTranslator
translator = BiopythonTranslator()
graphic_record = translator.translate_record("example_sequence.gb")
subrecord = graphic_record.crop((1700, 2000))
fig, axes = subrecord.plot_on_multiple_lines(
nucl_per_line=70, plot_sequence=True
)
fig.savefig("multiline_plot.png")

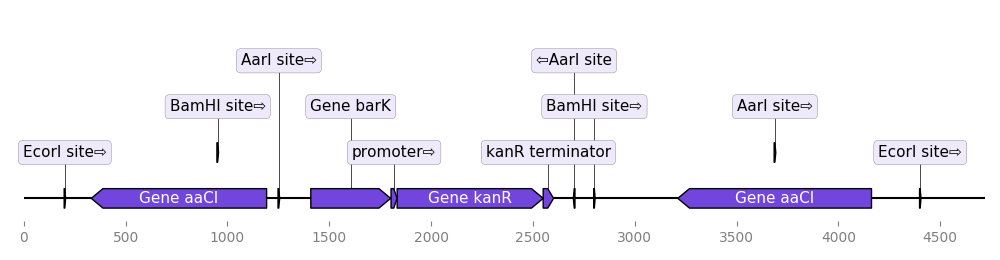
2.3.2.8. locally_highlighted_record¶
from dna_features_viewer import BiopythonTranslator
start, end = 1300, 2700
def feature_properties(f):
"""Fade away all features not overlapping with [start, end]"""
if f.location.end < start or f.location.start > end:
return dict(color="white", linecolor="grey", label=None)
return {}
translator = BiopythonTranslator(features_properties=feature_properties)
graphic_record = translator.translate_record("example_sequence.gb")
ax, _ = graphic_record.plot(figure_width=12, elevate_outline_annotations=True)
ax.fill_between(
[start, end], -10, 10, facecolor="peachpuff", alpha=0.2, zorder=-1
)
ax.figure.savefig('locally_highlighted_record.png', bbox_inches='tight')

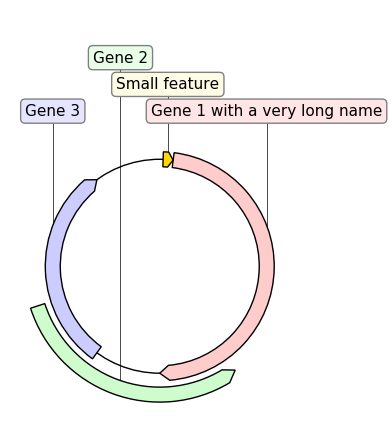
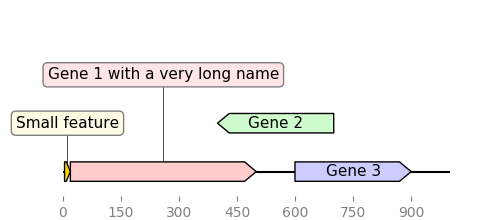
2.3.2.9. graphic_record_defined_by_hand¶
"""Simple example where a few features are defined "by hand" and are displayed
and exported as PNG, first with a linear view, then with a circular
view.
"""
from dna_features_viewer import (
GraphicFeature,
GraphicRecord,
CircularGraphicRecord,
)
features = [
GraphicFeature(
start=5, end=20, strand=+1, color="#ffd700", label="Small feature"
),
GraphicFeature(
start=20,
end=500,
strand=+1,
color="#ffcccc",
label="Gene 1 with a very long name",
),
GraphicFeature(
start=400, end=700, strand=-1, color="#cffccc", label="Gene 2"
),
GraphicFeature(
start=600, end=900, strand=+1, color="#ccccff", label="Gene 3"
),
]
# PLOT AND EXPORT A LINEAR VIEW OF THE CONSTRUCT
record = GraphicRecord(sequence_length=1000, features=features)
ax, _ = record.plot(figure_width=5)
ax.figure.savefig("graphic_record_defined_by_hand.png")
# PLOT AND EXPORT A CIRCULAR VIEW OF THE CONSTRUCT
circular_rec = CircularGraphicRecord(sequence_length=1000, features=features)
ax2, _ = circular_rec.plot(figure_width=4)
ax2.figure.tight_layout()
ax2.figure.savefig(
"graphic_record_defined_by_hand_circular.png", bbox_inches="tight"
)
2.3.2.10. gff_record_from_the_web¶
import urllib
from io import StringIO
from dna_features_viewer import BiopythonTranslator, load_record
# DEFINE FEATURES ASPECTS
def features_properties(f):
"""Mutations get a red label, other features get a pastel color."""
label = None
if f.type == "Mutagenesis":
label = f.qualifiers["Note"][0]
color = {
"Mutagenesis": "firebrick",
"Active site": "yellow",
"Beta strand": "lightyellow",
"Chain": "lightcyan",
"Helix": "honeydew",
"Initiator methionine": "white",
"Metal binding": "lightsteelblue",
"Turn": "moccasin",
}.get(f.type, "white")
return dict(color=color, label=label)
# GET THE RECORD FROM UNIPROT
response = urllib.request.urlopen("https://www.uniprot.org/uniprot/P0A7B8.gff")
record_file = StringIO(response.read().decode())
# TRANSLATE AND PLOT THE RECORD
translator = BiopythonTranslator(features_properties=features_properties)
graphic_record = translator.translate_record(record_file)
ax, _ = graphic_record.plot(
figure_width=15, max_label_length=100, elevate_outline_annotations=True,
)
ax.set_title("Mutation effects in P0A7B8", fontweight="bold", fontsize=16)
ax.figure.savefig("gff_record_from_the_web.png", bbox_inches="tight")

2.3.2.11. from_genbank¶
from dna_features_viewer import BiopythonTranslator
graphic_record = BiopythonTranslator().translate_record("example_sequence.gb")
ax, _ = graphic_record.plot(figure_width=10, strand_in_label_threshold=7)
ax.figure.tight_layout()
ax.figure.savefig("from_genbank.png")
2.3.2.12. example_with_inverted_x_axis¶
"""This example shows how you can very easily flip a plot horizontally if you
need, using Matplotlib's ax.set_xlim() method."""
from dna_features_viewer import BiopythonTranslator, load_record
ax = BiopythonTranslator.quick_class_plot("example_sequence.gb")
x1, x2 = ax.get_xlim()
ax.set_xlim(x2, x1)
ax.figure.tight_layout()
ax.figure.savefig("example_with_inverted_x_axis.png")

2.3.2.13. example_with_gif¶
"""An example with GIF generation at the end. How cool is that!
This example requires the Moviepy library installed (pip install moviepy).
"""
from Bio import Entrez, SeqIO
import moviepy.editor as mpe
from moviepy.video.io.bindings import mplfig_to_npimage
import matplotlib.pyplot as plt
from dna_features_viewer import BiopythonTranslator, CircularGraphicRecord
# DOWNLOAD THE PLASMID's RECORD FROM NCBI
handle = Entrez.efetch(
db="nucleotide", id=1473096477, rettype="gb", retmode="text"
)
record = SeqIO.read(handle, "genbank")
# CREATE THE GRAPHIC RECORD WITH DNA_FEATURES_VIEWER
color_map = {
"rep_origin": "yellow",
"CDS": "orange",
"regulatory": "red",
"misc_recomb": "darkblue",
"misc_feature": "lightblue",
}
translator = BiopythonTranslator(
features_filters=(lambda f: f.type not in ["gene", "source"],),
features_properties=lambda f: {"color": color_map.get(f.type, "white")},
)
translator.max_line_length = 15
graphic_record = translator.translate_record(
record, record_class=CircularGraphicRecord
)
graphic_record.labels_spacing = 15
# ANIMATE INTO A GIF WITH MOVIEPY
duration = 5
def make_frame(t):
top_nucleotide_index = t * graphic_record.sequence_length / duration
graphic_record.top_position = top_nucleotide_index
ax, _ = graphic_record.plot(figure_width=8, figure_height=11)
ax.set_ylim(top=2)
np_image = mplfig_to_npimage(ax.figure)
plt.close(ax.figure)
return np_image
clip = mpe.VideoClip(make_frame, duration=duration)
small_clip = clip.crop(x1=60, x2=-60, y1=100, y2=-100).resize(0.5)
small_clip.write_gif("example_with_gif.gif", fps=15)
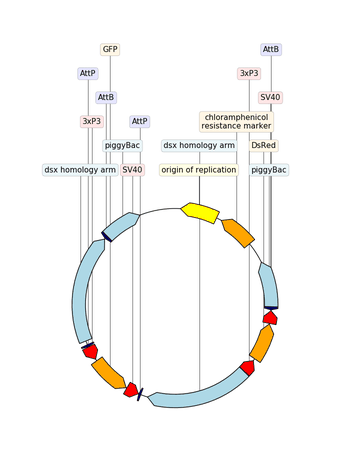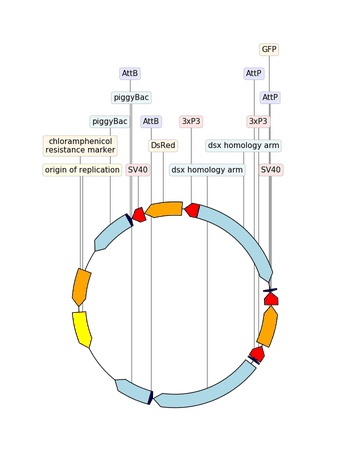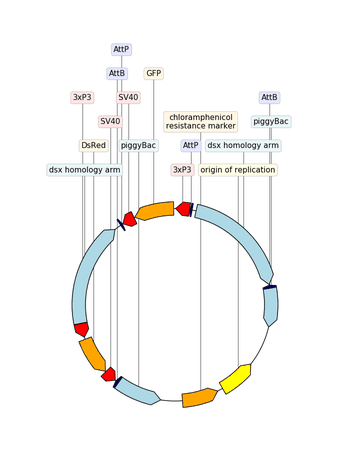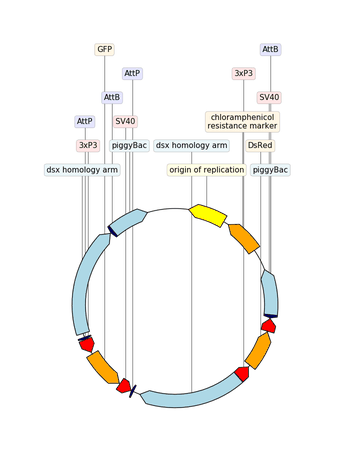
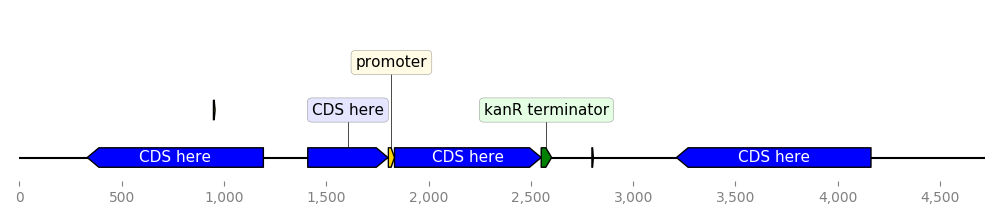
2.3.2.14. custom_biopython_translator¶
from dna_features_viewer import BiopythonTranslator
class MyCustomTranslator(BiopythonTranslator):
"""Custom translator implementing the following theme:
- Color terminators in green, CDS in blue, all other features in gold.
- Do not display features that are restriction sites unless they are BamHI
- Do not display labels for restriction sites.
- For CDS labels just write "CDS here" instead of the name of the gene.
"""
def compute_feature_color(self, feature):
if feature.type == "CDS":
return "blue"
elif feature.type == "terminator":
return "green"
else:
return "gold"
def compute_feature_label(self, feature):
if feature.type == 'restriction_site':
return None
elif feature.type == "CDS":
return "CDS here"
else:
return BiopythonTranslator.compute_feature_label(self, feature)
def compute_filtered_features(self, features):
"""Do not display promoters. Just because."""
return [
feature for feature in features
if (feature.type != "restriction_site")
or ("BamHI" in str(feature.qualifiers.get("label", '')))
]
graphic_record = MyCustomTranslator().translate_record("example_sequence.gb")
ax, _ = graphic_record.plot(figure_width=10)
ax.figure.tight_layout()
ax.figure.savefig("custom_biopython_translator.png")
2.3.2.15. cartoon_style¶
"""In this example, we draw features XKCD style.
We use Matplotlib's built-in xkcd() function, and a few tweaks:
- We set record.default_box_color to None to prevent annotations
box drawing.
- We set the record.default_font_family parameter for a nice font
for annotations
- We set plt.rcParams["font.family"] for a nice font for the
ruler.
"""
from matplotlib import rc_context
from dna_features_viewer import GraphicFeature, GraphicRecord
rc_context(
{
"font.family": ["Walter Turncoat"],
"path.sketch": (1.5, 300, 1), # scale, length, randomness
}
)
features = [
GraphicFeature(
start=20,
end=500,
strand=+1,
color="#ffcccc",
label="Gene 1 with a very, very long name",
box_linewidth=0,
box_color='white'
),
GraphicFeature(
start=400, end=700, strand=-1, color="#cffccc", label="Gene 2"
),
GraphicFeature(
start=600, end=900, strand=+1, color="#0000ff", label="Gene 3"
)
]
record = GraphicRecord(sequence_length=1000, features=features)
ax, _ = record.plot(figure_width=3)
ax.figure.tight_layout()
ax.figure.savefig("cartoon_style.png", dpi=200)

用户留言